
Characterization of an In Vivo Neutralizing Anti-Vaccinia Virus D8 Single-Chain Fragment Variable (scFv) from a Human Anti-Vaccinia Virus-Specific Recombinant Library

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Immunization and Lymphocyte Preparation
2.2. Library Construction
2.3. Cells and Viruses
2.4. Gradient Purification of Vaccinia Viruses Elstree and Munich 1
2.5. ScFv Selection Using Purified Vaccinia Virus Elstree
2.6. Screening of Randomly Selected scFv-Producing HB2151 Clones in Indirect ELISAs
2.7. Purification of Selected scFvs
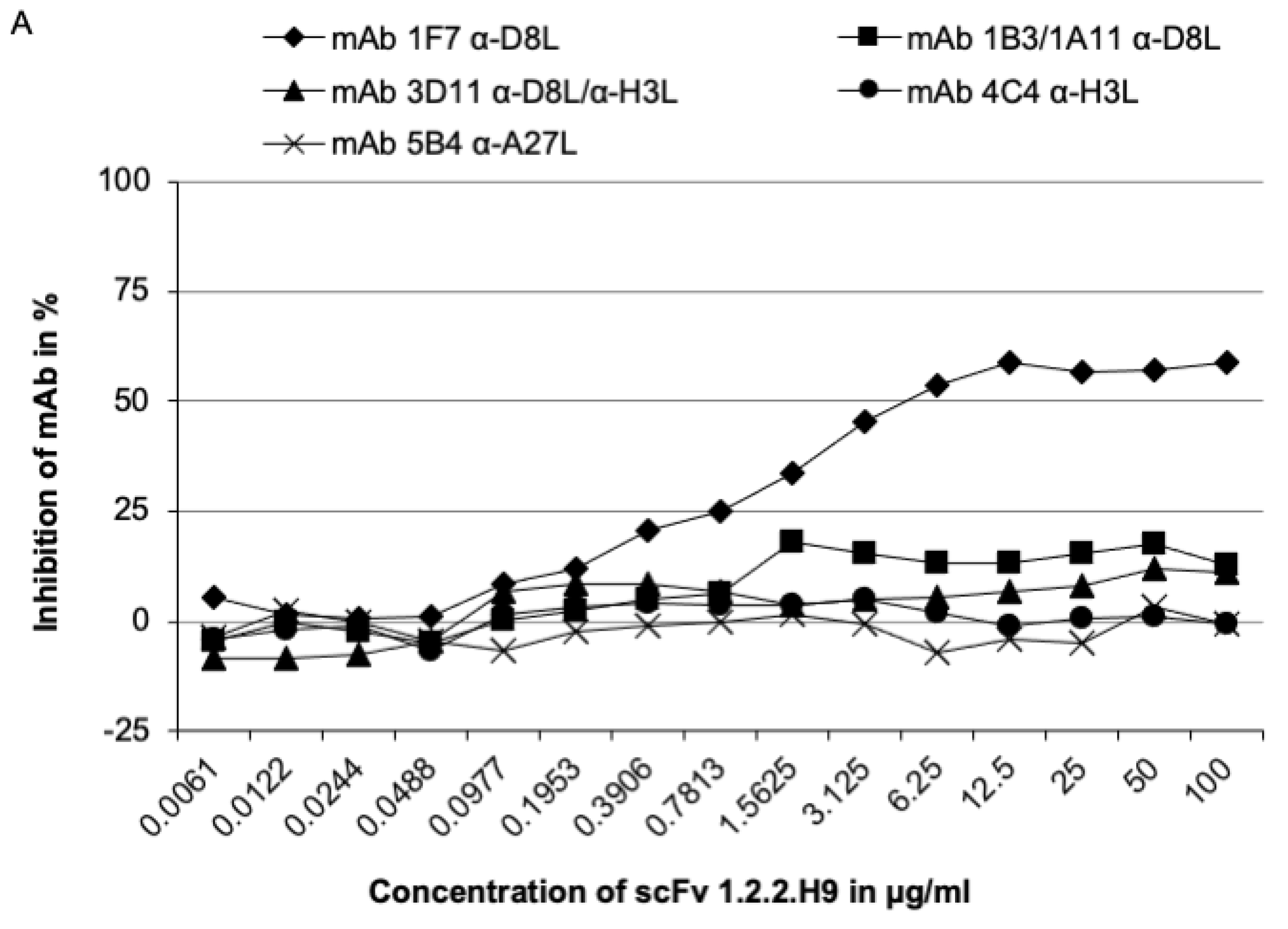
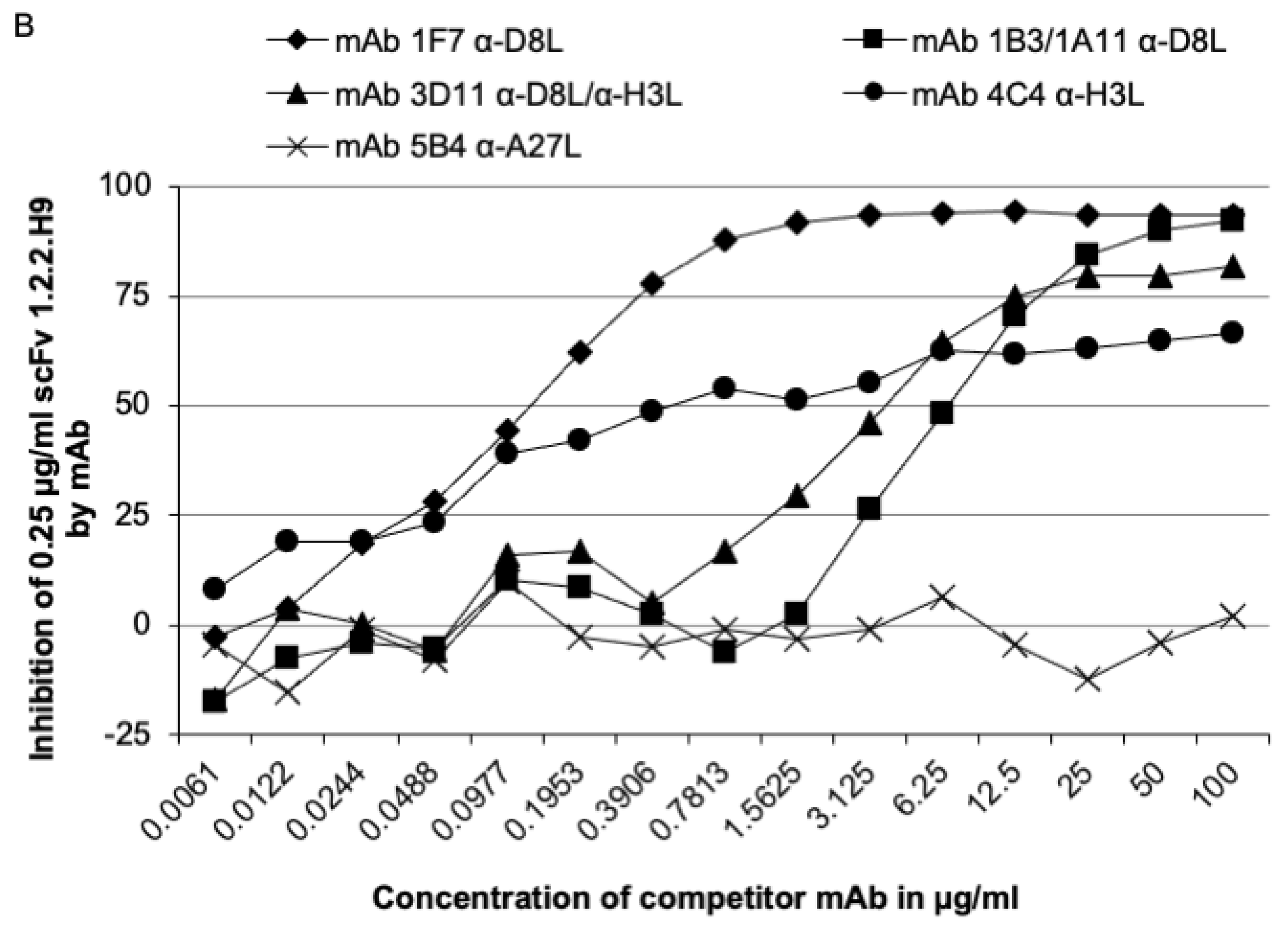
2.8. Competitive ELISA for Epitope Detection
2.9. Engineering of Specific Binding scFv to Human scFv-Fc and IgG1 Molecules
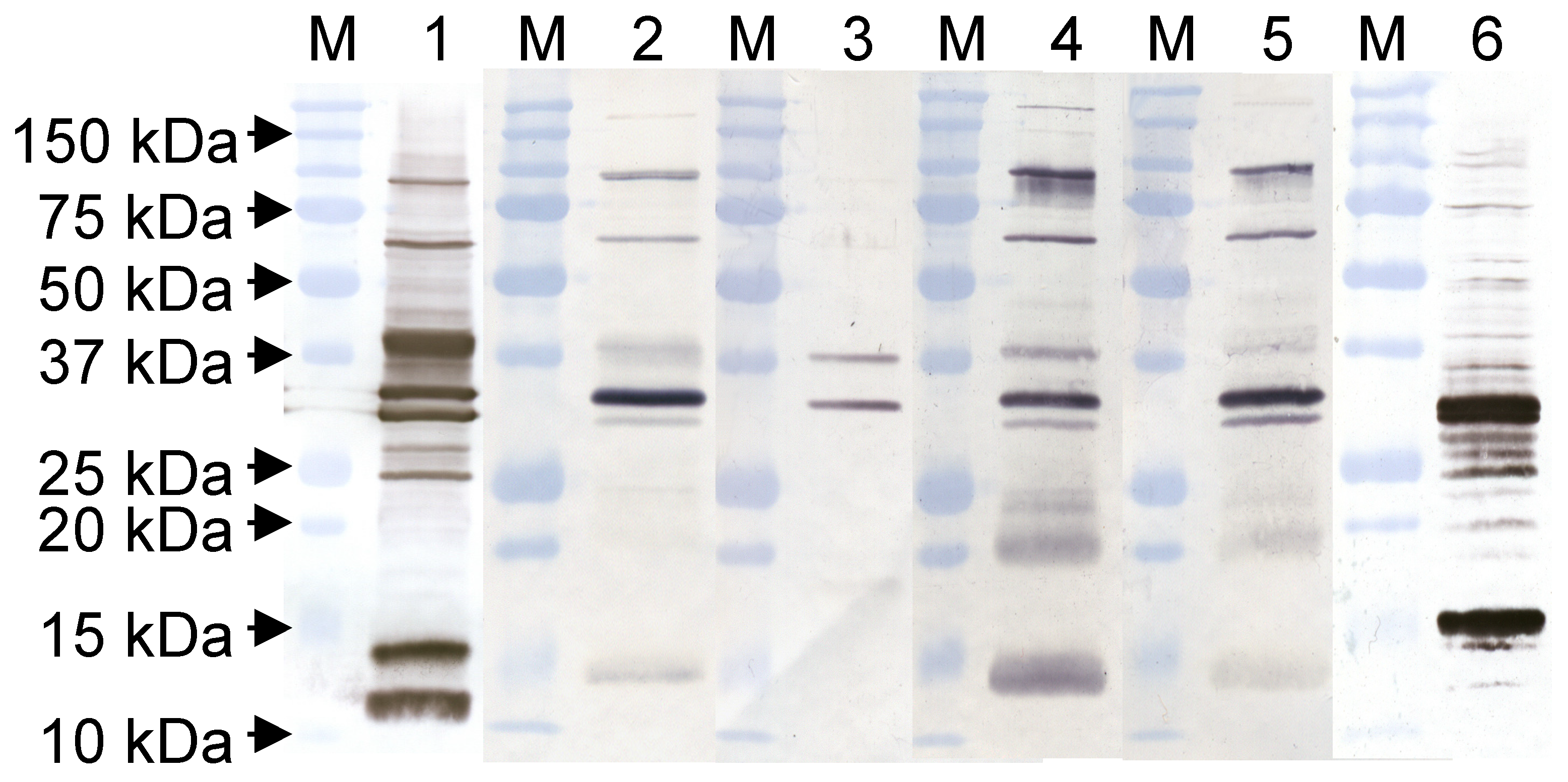
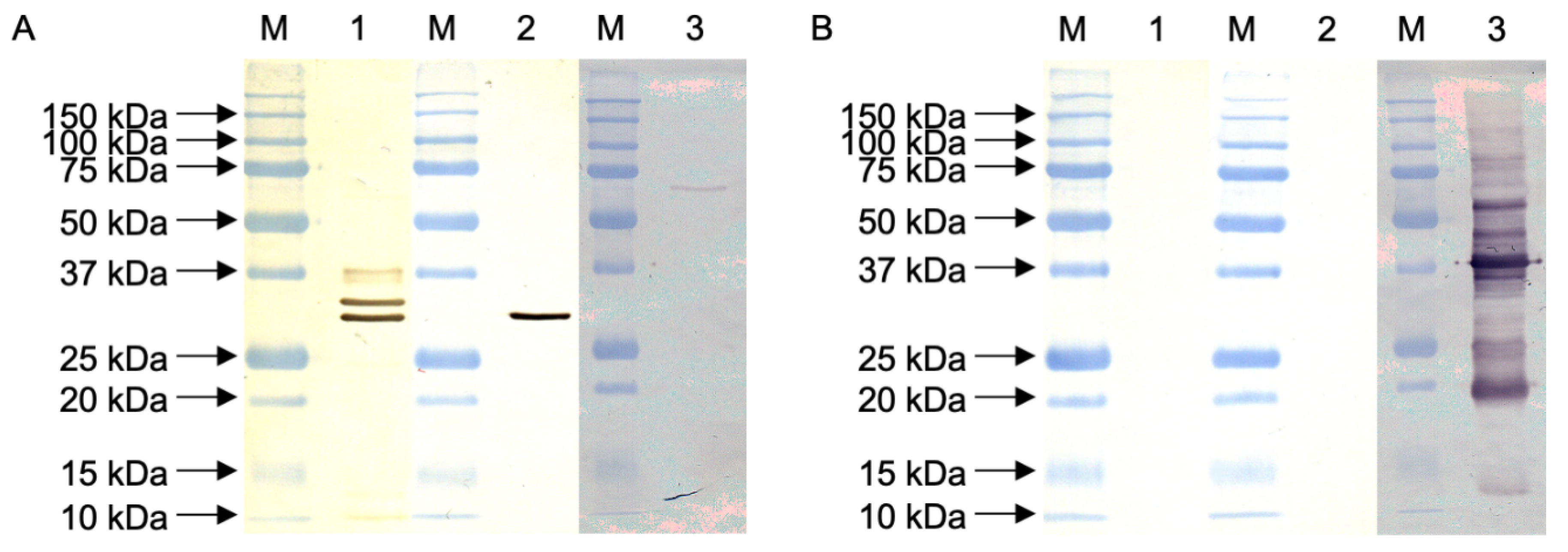
2.10. SDS-PAGE and Western Blotting for the Detection of the Target Virus Protein
2.11. Enzyme-Linked Immunosorbent Assay (ELISA)
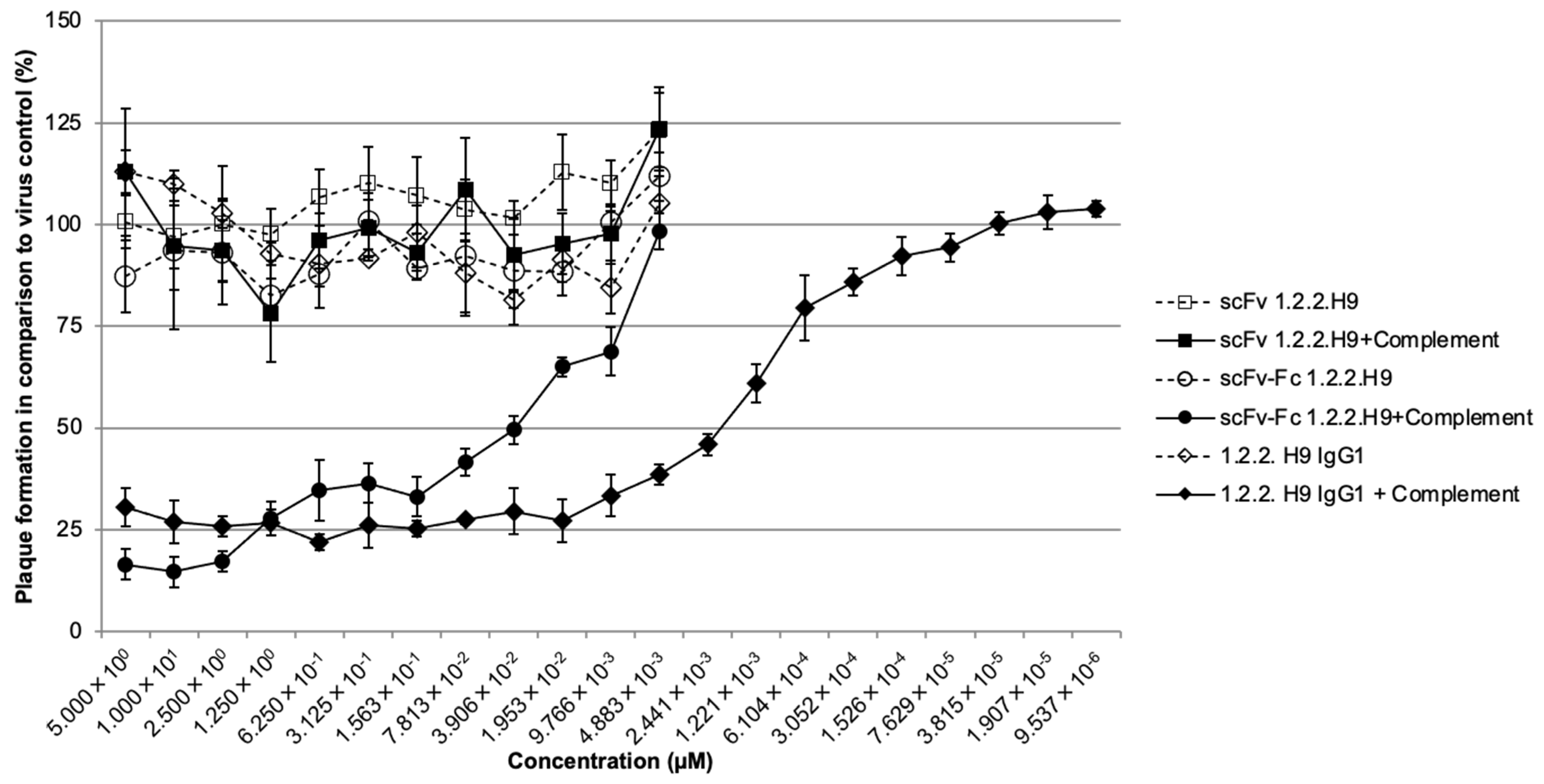
2.12. In Vitro Plaque Reduction Neutralization Test (PRNT)
2.13. In Vivo Neutralization
3. Results
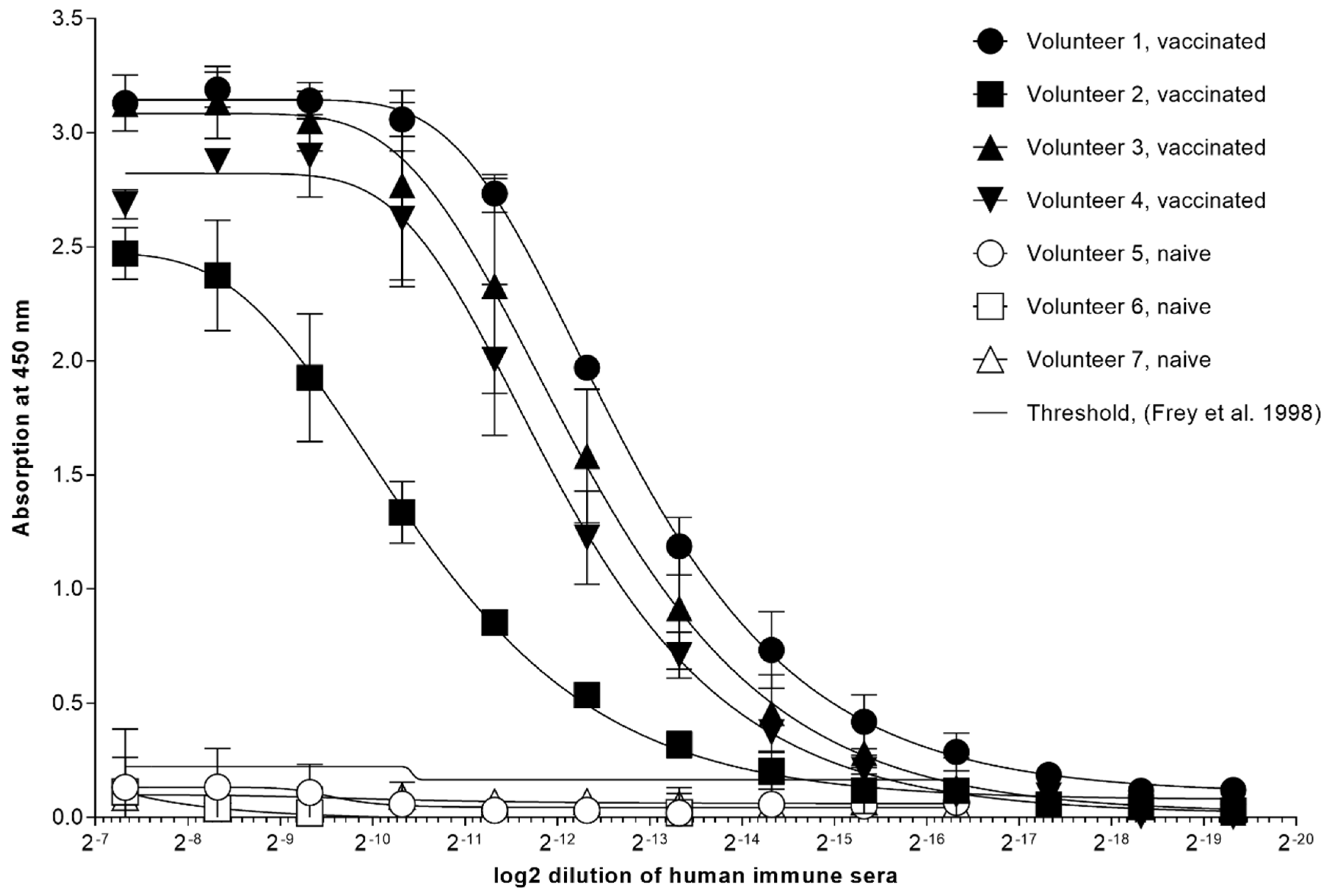
3.1. Immunization, Library Construction, and Characterization
3.2. Selection of Vaccinia-Virus-Specific scFv
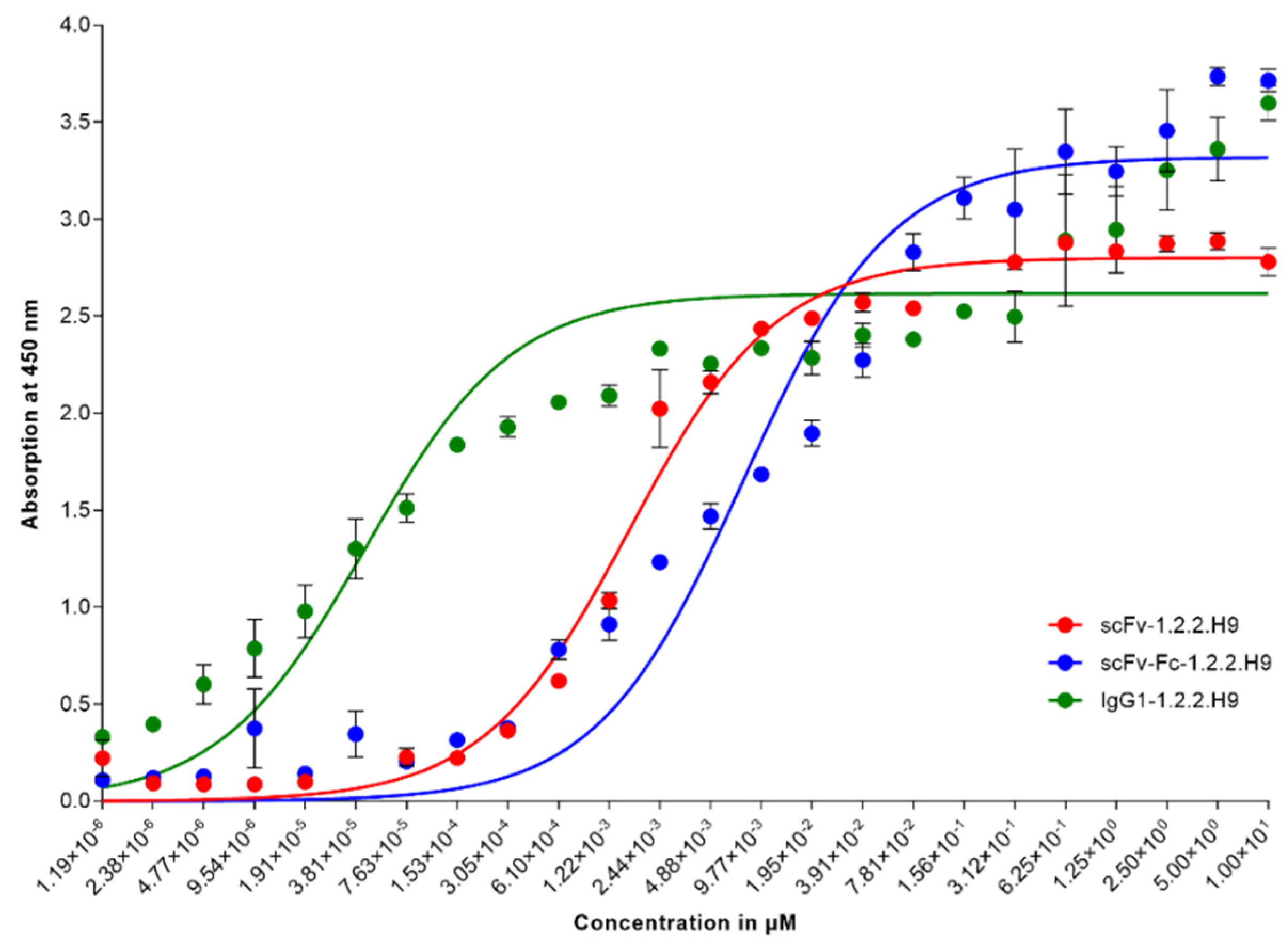
3.3. Binding Characteristics of scFv-1.2.2.H9, scFv-Fc-1.2.2.H9, and IgG1-1.2.2.H9 in ELISA
3.4. Epitope Mapping
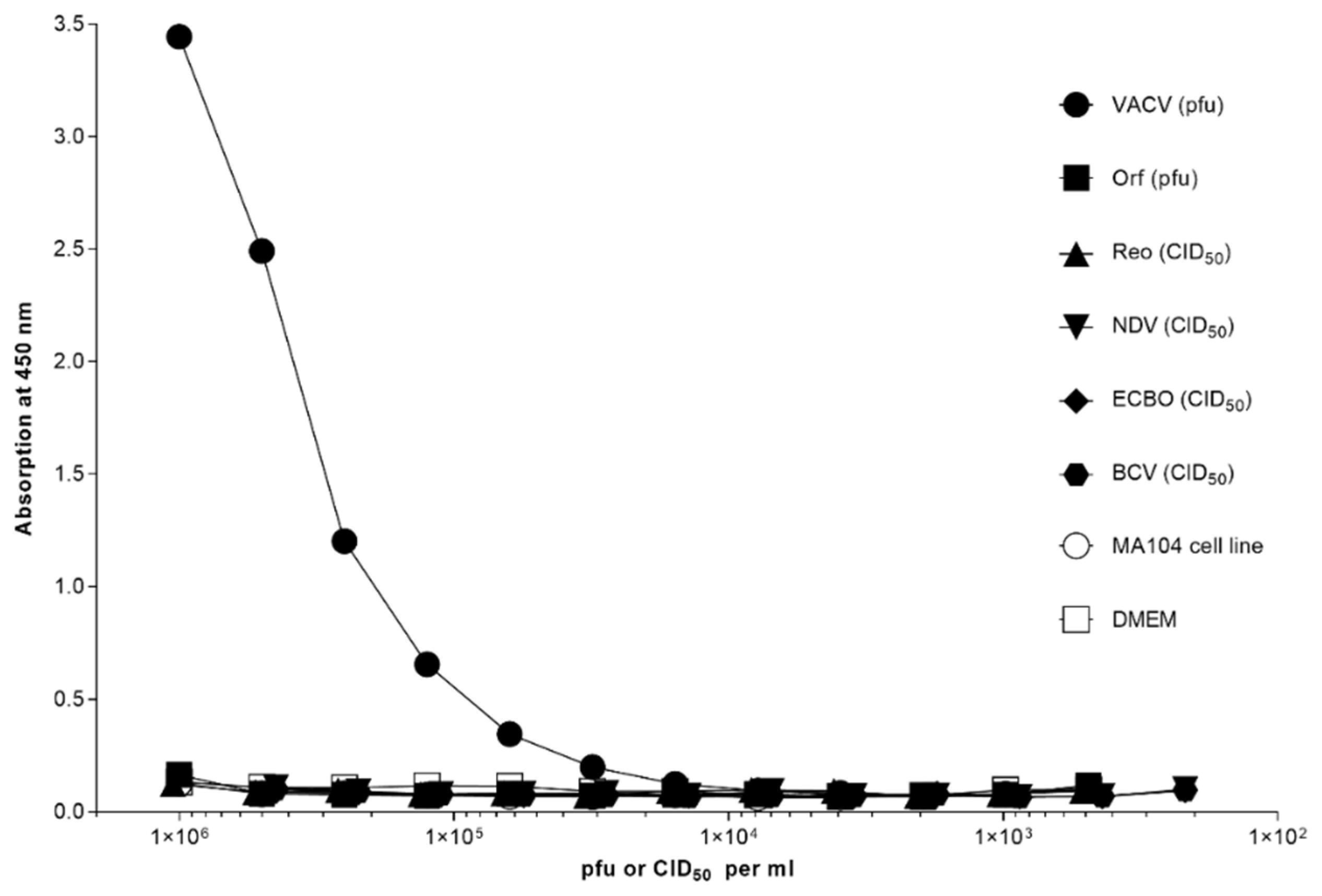
3.5. In Vitro Neutralization
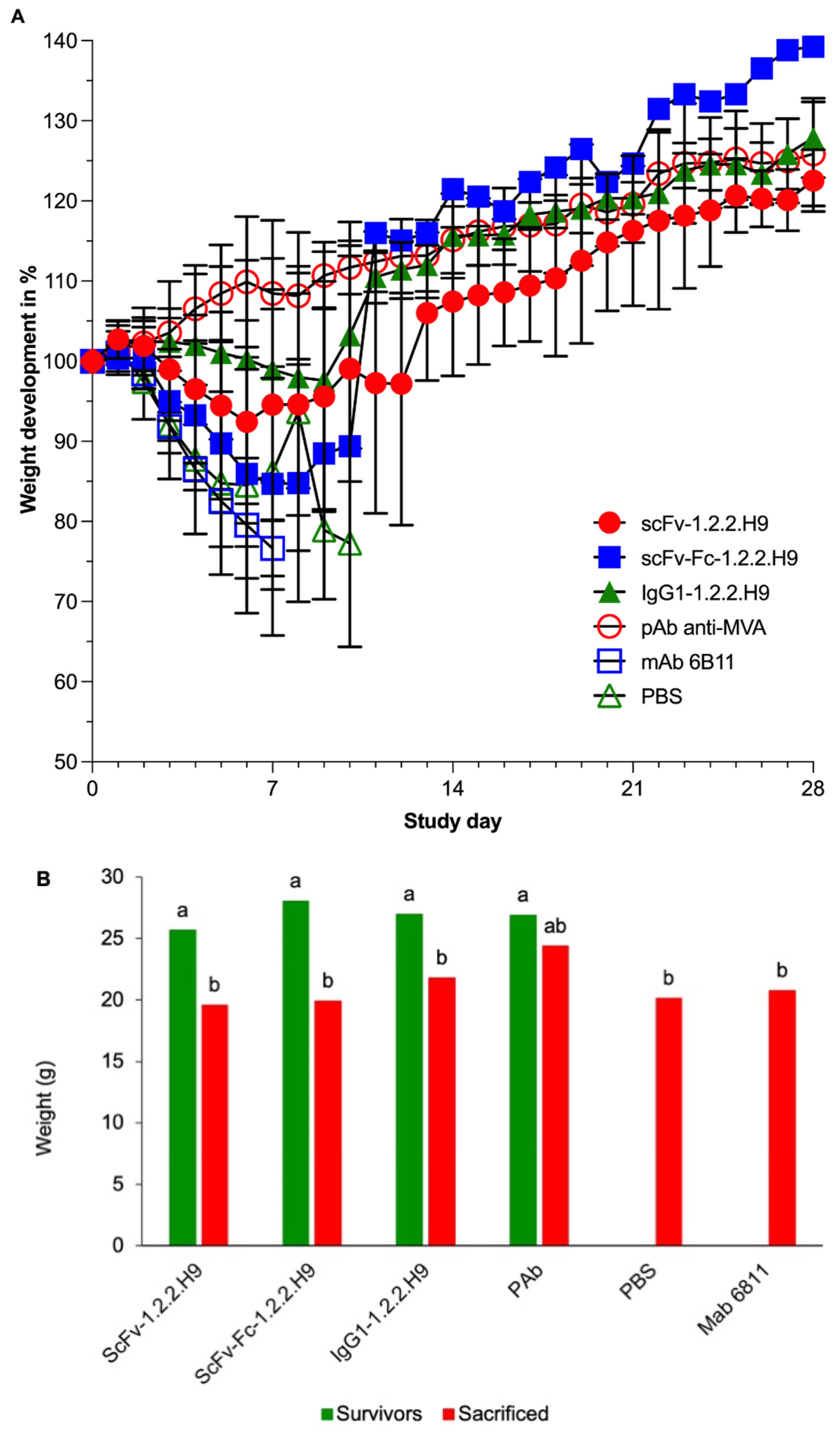
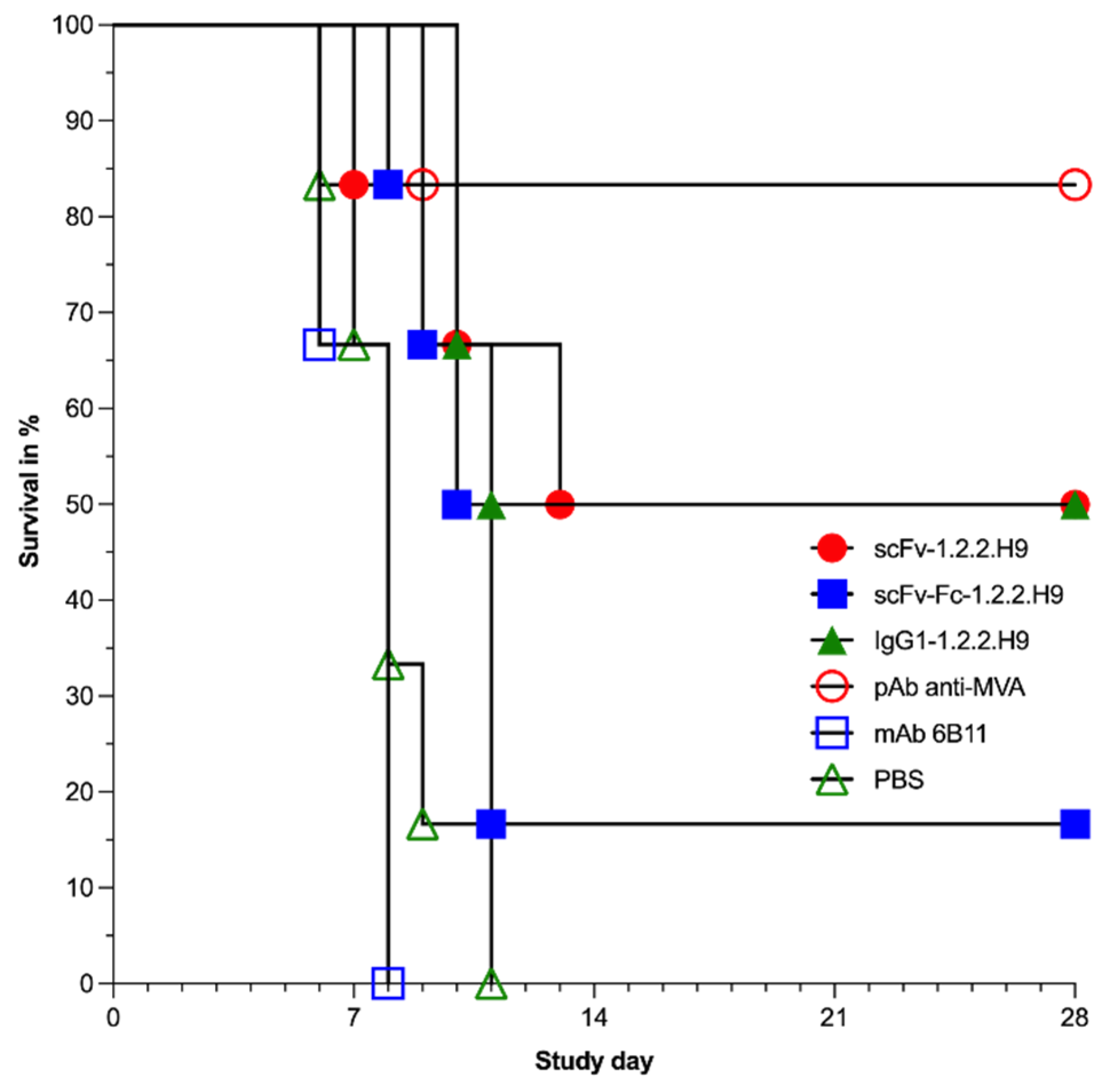
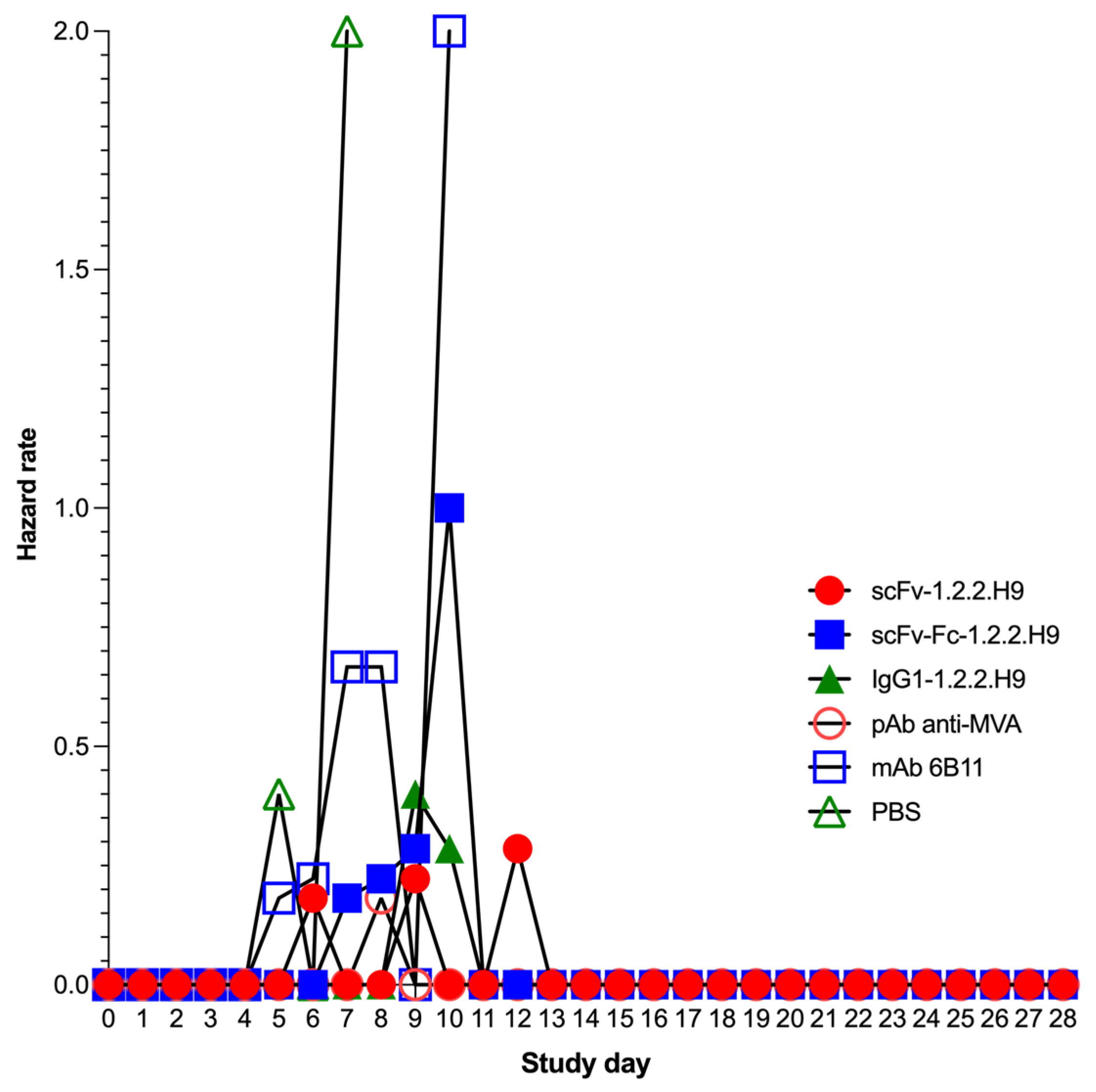
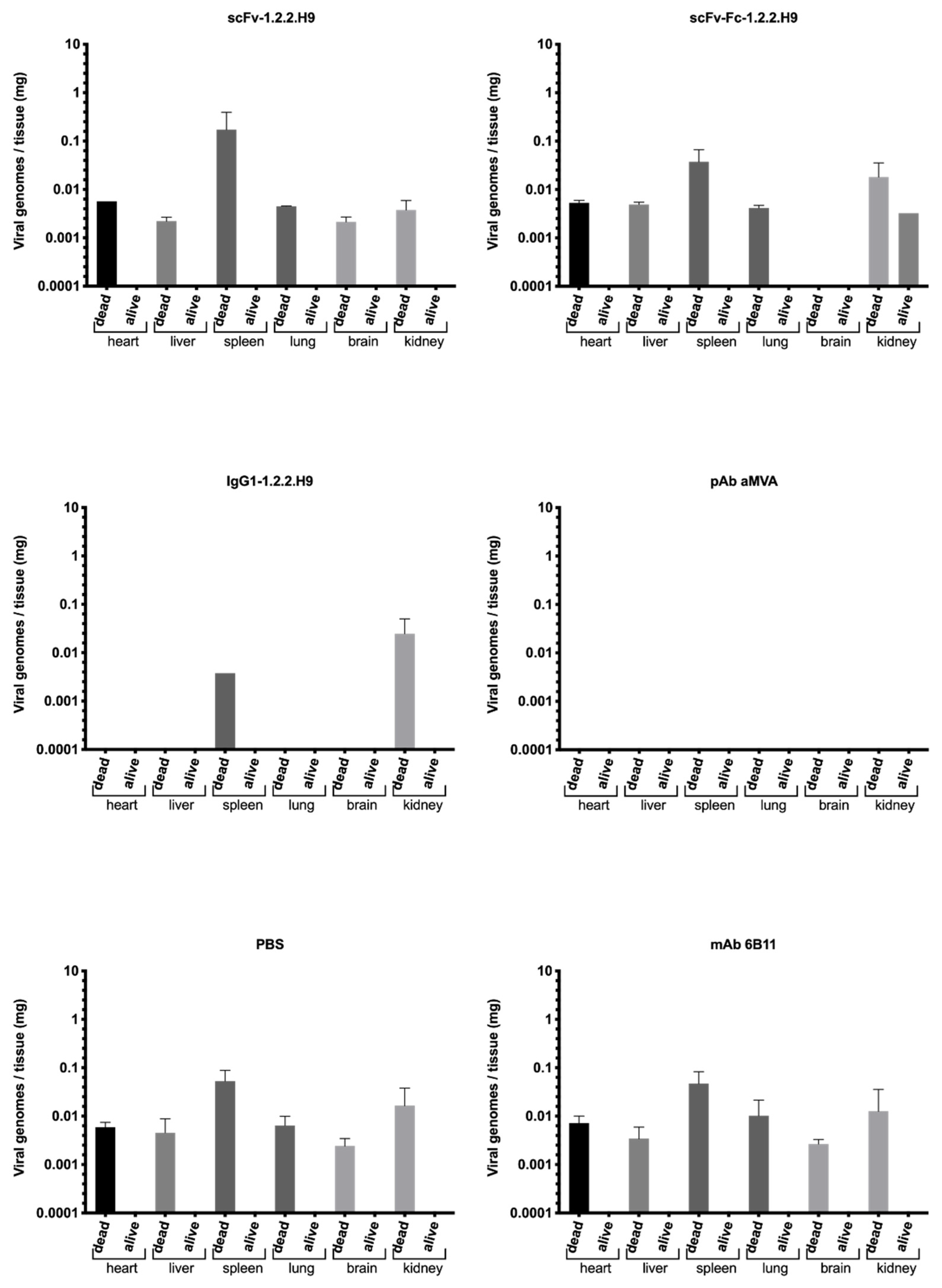
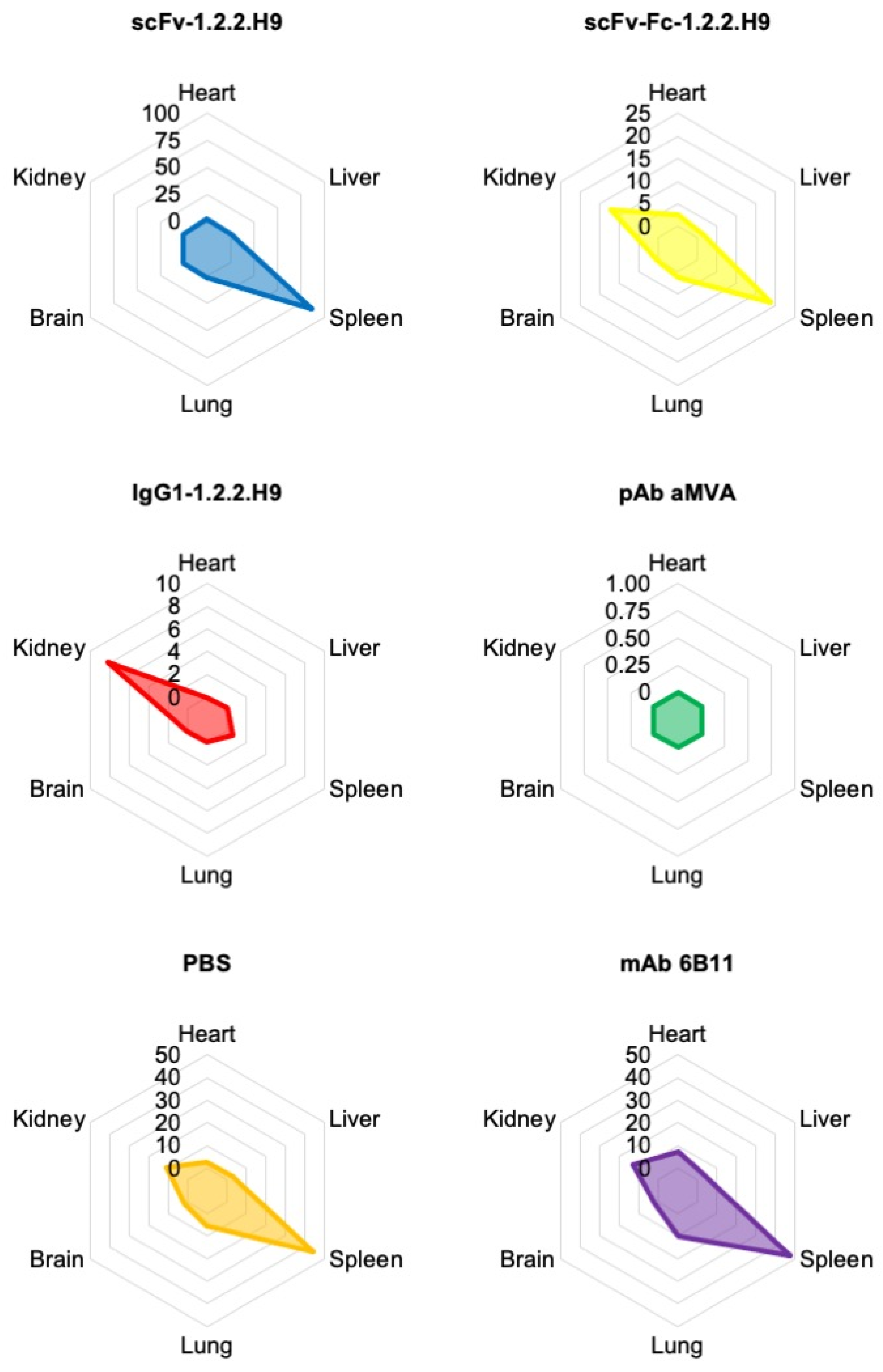
3.6. In Vivo Passive Protection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moss, B. Poxvirus entry and membrane fusion. Virology 2006, 344, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D. Smallpox and Its Eradication; World Helth Organization: Geneva, Switzerland, 1988; pp. 1–1460. [Google Scholar]
- Ladnyi, I.D.; Breman, J.G. Smallpox eradication: Progress and problems. Dev. Biol. Stand. 1978, 41, 281–290. [Google Scholar]
- Kurth, A.; Wibbelt, G.; Gerber, H.P.; Petschaelis, A.; Pauli, G.; Nitsche, A. Rat-to-elephant-to-human transmission of cowpox virus. Emerg. Infect. Dis. 2008, 14, 670–671. [Google Scholar] [CrossRef]
- Becker, C.; Kurth, A.; Hessler, F.; Kramp, H.; Gokel, M.; Hoffmann, R.; Kuczka, A.; Nitsche, A. Cowpox virus infection in pet rat owners: Not always immediately recognized. Dtsch. Arztebl. Int. 2009, 106, 329–334. [Google Scholar] [CrossRef]
- Vorou, R.M.; Papavassiliou, V.G.; Pierroutsakos, I.N. Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 2008, 21, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Campe, H.; Zimmermann, P.; Glos, K.; Bayer, M.; Bergemann, H.; Dreweck, C.; Graf, P.; Weber, B.K.; Meyer, H.; Büttner, M.; et al. Cowpox virus transmission from pet rats to humans, Germany. Emerg. Infect. Dis. 2009, 15, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Ladnyj, I.D.; Ziegler, P.; Kima, E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull. World Health Organ. 1972, 46, 593–597. [Google Scholar]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The detection of monkeypox in humans in the Western Hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, A.; Aarons, E.; Astbury, J.; Balasegaram, S.; Beadsworth, M.; Beck, C.R.; Chand, M.; O’Connor, C.; Dunning, J.; Ghebrehewet, S.; et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Eurosurveill 2018, 23, 1800509. [Google Scholar] [CrossRef] [PubMed]
- Fulginiti, V.A. Risks of smallpox vaccination. JAMA 2003, 290, 1452. [Google Scholar] [PubMed] [Green Version]
- Cono, J.; Casey, C.G.; Bell, D.M. Smallpox vaccination and adverse reactions. Guidance for clinicians. MMWR Recomm. Rep. 2003, 52, 1–28. [Google Scholar] [PubMed]
- Fulginiti, V.A.; Papier, A.; Lane, J.M.; Neff, J.M.; Henderson, D.A. Smallpox vaccination: A review, part II. Adverse events. Clin. Infect. Dis. 2003, 37, 251–271. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, R.J.; Lane, J.M. Clinical efficacy of intramuscular vaccinia immune globulin: A literature review. Clin. Infect. Dis. 2004, 39, 819–826. [Google Scholar] [CrossRef]
- Hopkins, R.J.; Kramer, W.G.; Blackwelder, W.C.; Ashtekar, M.; Hague, L.; Winker-La Roche, S.D.; Berezuk, G.; Smith, D.; Leese, P.T. Safety and pharmacokinetic evaluation of intravenous vaccinia immune globulin in healthy volunteers. Clin. Infect. Dis. 2004, 39, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Kempe, C.H. Studies smallpox and complications of smallpox vaccination. Pediatrics 1960, 26, 176–189. [Google Scholar]
- Feery, B.J. The efficacy of vaccinial immune globulin. A 15-year study. Vox Sang. 1976, 31, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, L.A. Antibodies for the prevention and treatment of viral diseases. Antivir. Res. 2000, 47, 57–77. [Google Scholar] [CrossRef]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 2002, 83, 2915–2931. [Google Scholar] [CrossRef]
- Boulter, E.A.; Appleyard, G. Differences between extracellular and intracellular forms of poxvirus and their implications. Prog. Med. Virol. 1973, 16, 86–108. [Google Scholar]
- Blasco, R.; Moss, B. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J. Virol. 1992, 66, 4170–4179. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, J.C.; Chung, C.S.; Chang, W. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 1999, 73, 8750–8761. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Chung, C.S.; Heine, H.G.; Chang, W. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 2000, 74, 3353–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.F.; Janeczko, R.; Esteban, M. Isolation and characterization of neutralizing monoclonal antibodies to vaccinia virus. J. Virol. 1985, 56, 482–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallengren, K.; Risco, C.; Krijnse-Locker, J.; Esteban, M.; Rodriguez, D. The A17L gene product of vaccinia virus is exposed on the surface of IMV. Virology 2001, 290, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Wolffe, E.J.; Vijaya, S.; Moss, B. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 1995, 211, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Ichihashi, Y.; Oie, M. Neutralizing epitope on penetration protein of vaccinia virus. Virology 1996, 220, 491–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhnia, M.R.; McCausland, M.M.; Su, H.P.; Singh, K.; Hoffmann, J.; Davies, D.H.; Felgner, P.L.; Head, S.; Sette, A.; Garboczi, D.N.; et al. Redundancy and plasticity of neutralizing antibody responses are cornerstone attributes of the human immune response to the smallpox vaccine. J. Virol. 2008, 82, 3751–3768. [Google Scholar] [CrossRef] [Green Version]
- Eto, A.; Fujita, M.; Nishiyama, Y.; Saito, T.; Molina, D.M.; Morikawa, S.; Saijo, M.; Shinmura, Y.; Kanatani, Y. Profiling of the antibody response to attenuated LC16m8 smallpox vaccine using protein array analysis. Vaccine 2019, 37, 6588–6593. [Google Scholar] [CrossRef]
- Xu, C.; Meng, X.; Yan, B.; Crotty, S.; Deng, J.; Xiang, Y. An epitope conserved in orthopoxvirus A13 envelope protein is the target of neutralizing and protective antibodies. Virology 2011, 418, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Ahsendorf, H.P.; Gan, L.L.; Eltom, K.H.; Abd El Wahed, A.; Hotop, S.K.; Roper, R.L.; Beutling, U.; Broenstrup, M.; Stahl-Hennig, C.; Hoelzle, L.E.; et al. Species-Specific Conservation of Linear Antigenic Sites on Vaccinia Virus A27 Protein Homologs of Orthopoxviruses. Viruses 2019, 11, 493. [Google Scholar] [CrossRef] [Green Version]
- Kaever, T.; Matho, M.H.; Meng, X.; Crickard, L.; Schlossman, A.; Xiang, Y.; Crotty, S.; Peters, B.; Zajonc, D.M. Linear Epitopes in Vaccinia Virus A27 Are Targets of Protective Antibodies Induced by Vaccination against Smallpox. J. Virol. 2016, 90, 4334–4345. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Earl, P.; Americo, J.; Damon, I.; Smith, S.K.; Zhou, Y.H.; Yu, F.; Sebrell, A.; Emerson, S.; Cohen, G.; et al. Chimpanzee/human mAbs to vaccinia virus B5 protein neutralize vaccinia and smallpox viruses and protect mice against vaccinia virus. Proc. Natl. Acad. Sci. USA 2006, 103, 1882–1887. [Google Scholar] [CrossRef] [Green Version]
- Aldaz-Carroll, L.; Whitbeck, J.C.; Ponce de Leon, M.; Lou, H.; Hirao, L.; Isaacs, S.N.; Moss, B.; Eisenberg, R.J.; Cohen, G.H. Epitope-mapping studies define two major neutralization sites on the vaccinia virus extracellular enveloped virus glycoprotein B5R. J. Virol. 2005, 79, 6260–6271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czerny, C.P.; Johann, S.; Holzle, L.; Meyer, H. Epitope detection in the envelope of intracellular naked orthopox viruses and identification of encoding genes. Virology 1994, 200, 764–777. [Google Scholar] [CrossRef]
- Matho, M.H.; Maybeno, M.; Benhnia, M.R.; Becker, D.; Meng, X.; Xiang, Y.; Crotty, S.; Peters, B.; Zajonc, D.M. Structural and biochemical characterization of the vaccinia virus envelope protein D8 and its recognition by the antibody LA5. J. Virol. 2012, 86, 8050–8058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilchuk, I.; Gilchuk, P.; Sapparapu, G.; Lampley, R.; Singh, V.; Kose, N.; Blum, D.L.; Hughes, L.J.; Satheshkumar, P.S.; Townsend, M.B.; et al. Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections. Cell 2016, 167, 684–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maa, J.S.; Rodriguez, J.F.; Esteban, M. Structural and functional characterization of a cell surface binding protein of vaccinia virus. J. Biol. Chem. 1990, 265, 1569–1577. [Google Scholar] [CrossRef]
- Matho, M.H.; de Val, N.; Miller, G.M.; Brown, J.; Schlossman, A.; Meng, X.; Crotty, S.; Peters, B.; Xiang, Y.; Hsieh-Wilson, L.C.; et al. Murine anti-vaccinia virus D8 antibodies target different epitopes and differ in their ability to block D8 binding to CS-E. PLoS Pathog. 2014, 10, e1004495. [Google Scholar] [CrossRef] [Green Version]
- Mirzakhanyan, Y.; Gershon, P. The Vaccinia virion: Filling the gap between atomic and ultrastructure. PLoS Pathog. 2019, 15, e1007508. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.R.; Rodriguez, D.; Esteban, M. Insertional inactivation of the vaccinia virus 32-kilodalton gene is associated with attenuation in mice and reduction of viral gene expression in polarized epithelial cells. J. Virol. 1992, 66, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Niles, E.G.; Seto, J. Vaccinia virus gene D8 encodes a virion transmembrane protein. J. Virol. 1988, 62, 3772–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakhatskyy, P.; Wang, S.; Chou, T.H.; Lu, S. Immunogenicity and protection efficacy of monovalent and polyvalent poxvirus vaccines that include the D8 antigen. Virology 2006, 355, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Matho, M.H.; Schlossman, A.; Gilchuk, I.M.; Miller, G.; Mikulski, Z.; Hupfer, M.; Wang, J.; Bitra, A.; Meng, X.; Xiang, Y.; et al. Structure-function characterization of three human antibodies targeting the vaccinia virus adhesion molecule D8. J. Biol. Chem. 2018, 293, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogenboom, H.R.; de Bruine, A.P.; Hufton, S.E.; Hoet, R.M.; Arends, J.W.; Roovers, R.C. Antibody phage display technology and its applications. Immunotechnology 1998, 4, 1–20. [Google Scholar] [CrossRef]
- Schmaljohn, C.; Cui, Y.; Kerby, S.; Pennock, D.; Spik, K. Production and characterization of human monoclonal antibody Fab fragments to vaccinia virus from a phage-display combinatorial library. Virology 1999, 258, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.; Di Canzio, J.; Zurakowski, D. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods 1998, 221, 35–41. [Google Scholar] [CrossRef]
- Hurteau, G.J.; Spivack, S.D. mRNA-specific reverse transcription-polymerase chain reaction from human tissue extracts. Anal. Biochem. 2002, 307, 304–315. [Google Scholar] [CrossRef]
- Czerny, C.P.; Mahnel, H. Structural and functional analysis of orthopoxvirus epitopes with neutralizing monoclonal antibodies. J. Gen. Virol. 1990, 71, 2341–2352. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Czerny, C.P.; Waldmann, R.; Scheubeck, T. Identification of three distinct antigenic sites in parapoxviruses. Arch. Virol. 1997, 142, 807–821. [Google Scholar] [CrossRef]
- Brochet, X.; Lefranc, M.P.; Giudicelli, V. IMGT/V-QUEST: The highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008, 36, W503–W508. [Google Scholar] [CrossRef] [Green Version]
- Giudicelli, V.; Brochet, X.; Lefranc, M.P. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc. 2011, 2011, 695–715. [Google Scholar] [CrossRef]
- Steinwand, M.; Droste, P.; Frenzel, A.; Hust, M.; Dubel, S.; Schirrmann, T. The influence of antibody fragment format on phage display based affinity maturation of IgG. mAbs 2013, 6, 204–218. [Google Scholar] [CrossRef] [Green Version]
- Durocher, Y.; Perret, S.; Kamen, A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, E9. [Google Scholar] [CrossRef]
- Jager, V.; Bussow, K.; Wagner, A.; Weber, S.; Hust, M.; Frenzel, A.; Schirrmann, T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotech. 2013, 13, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M.L. Die Kinetik der Invertinwirkung. Biochem. Z. 1913, 49, 333–369. [Google Scholar]
- Czerny, C.P.; Mahnel, H.; Hornstein, O. Testing the immunity to orthopoxviruses in the white mouse with vaccinia virus. Zent. Vet. B 1989, 36, 100–112. [Google Scholar]
- Czerny, C.-P.; Alex, M.; Pricelius, J.; Zeller-Lue, C. Development of quantitative PCR tests for the detection of the orthopox virus adsorption protein gene (ORF D8L) on the Light CyclerTM. In Rapid Real Time PCR—A Light Cycler Application; Meuer, S., Wittwer, C., Eds.; Springer: New York, NY, USA, 2001; pp. 371–379. [Google Scholar]
- Smith, G.L.; Vanderplasschen, A. Extracellular enveloped vaccinia virus. Entry, egress, and evasion. Adv. Exp. Med. Biol. 1998, 440, 395–414. [Google Scholar]
- McCausland, M.M.; Benhnia, M.R.; Crickard, L.; Laudenslager, J.; Granger, S.W.; Tahara, T.; Kubo, R.; Koriazova, L.; Kato, S.; Crotty, S. Combination therapy of vaccinia virus infection with human anti-H3 and anti-B5 monoclonal antibodies in a small animal model. Antivir. Ther. 2010, 15, 661–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhnia, M.R.; McCausland, M.M.; Laudenslager, J.; Granger, S.W.; Rickert, S.; Koriazova, L.; Tahara, T.; Kubo, R.T.; Kato, S.; Crotty, S. Heavily isotype-dependent protective activities of human antibodies against vaccinia virus extracellular virion antigen B5. J. Virol. 2009, 83, 12355–12367. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.C.; Tapia, E.; Esteban, M. Administration to mice of a monoclonal antibody that neutralizes the intracellular mature virus form of vaccinia virus limits virus replication efficiently under prophylactic and therapeutic conditions. J. Gen. Virol. 2002, 83, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.R.; Winter, G. The binding site for C1q on IgG. Nature 1988, 332, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Benhnia, M.R.; McCausland, M.M.; Moyron, J.; Laudenslager, J.; Granger, S.; Rickert, S.; Koriazova, L.; Kubo, R.; Kato, S.; Crotty, S. Vaccinia virus extracellular enveloped virion neutralization in vitro and protection in vivo depend on complement. J. Virol. 2009, 83, 1201–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slifka, M.K.; Matloubian, M.; Ahmed, R. Bone marrow is a major site of long-term antibody production after acute viral infection. J. Virol. 1995, 69, 1895–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epitope ID | MAb | Isotype | Virus Strain Used for mAb Production | Isotype |
|---|---|---|---|---|
| 2A | 1B3/1A11 | IgG2a | VACV M1 | IgG2a |
| 2B | 1F7/2F9 | IgG2b | ECTV M1 | IgG2b |
| 2D | 3D11/2G7 | IgG2a | CPXV KR2 Brighton | IgG1 |
| 2G | 4C4/2B6 | IgG2a | CPXV KR2 Brighton | IgG1 |
| Equality Test over Levels | ||||
|---|---|---|---|---|
| Test | Chi-Quadrat | DF | Pr > Chi-Quadrat | |
| Log-Rank | 20.8477 | 5 | 0.0009 | |
| Adjustment for Multiple Comparisons for the Log-Rank Test | ||||
| Strata Comparison | Chi-Quadrat | p-Values | ||
| Group | Group | Raw | Sidak Adjustment | |
| 1 | 2 | 1.2919 | 0.2557 | 0.9881 |
| 1 | 3 | 0.00968 | 0.9216 | 1.0000 |
| 1 | 4 | 0.8085 | 0.3686 | 0.9990 |
| 1 | 5 | 2.1580 | 0.1418 | 0.8992 |
| 1 | 6 | 6.8927 | 0.0087 | 0.1222 |
| 2 | 3 | 1.4918 | 0.2219 | 0.9768 |
| 2 | 4 | 4.2725 | 0.0387 | 0.4471 |
| 2 | 5 | 0.1233 | 0.7255 | 1.0000 |
| 2 | 6 | 1.9182 | 0.1661 | 0.9344 |
| 3 | 4 | 0.6326 | 0.4264 | 0.9998 |
| 3 | 5 | 2.3846 | 0.1225 | 0.8593 |
| 3 | 6 | 7.3013 | 0.0069 | 0.0985 |
| 4 | 5 | 5.7236 | 0.0167 | 0.2237 |
| 4 | 6 | 13.4020 | 0.0003 | 0.0038 |
| 5 | 6 | 0.9339 | 0.3339 | 0.9977 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diesterbeck, U.S.; Ahsendorf, H.P.; Frenzel, A.; Sharifi, A.R.; Schirrmann, T.; Czerny, C.-P. Characterization of an In Vivo Neutralizing Anti-Vaccinia Virus D8 Single-Chain Fragment Variable (scFv) from a Human Anti-Vaccinia Virus-Specific Recombinant Library. Vaccines 2021, 9, 1308. https://doi.org/10.3390/vaccines9111308
Diesterbeck US, Ahsendorf HP, Frenzel A, Sharifi AR, Schirrmann T, Czerny C-P. Characterization of an In Vivo Neutralizing Anti-Vaccinia Virus D8 Single-Chain Fragment Variable (scFv) from a Human Anti-Vaccinia Virus-Specific Recombinant Library. Vaccines. 2021; 9(11):1308. https://doi.org/10.3390/vaccines9111308
Chicago/Turabian StyleDiesterbeck, Ulrike S., Henrike P. Ahsendorf, André Frenzel, Ahmad Reza Sharifi, Thomas Schirrmann, and Claus-Peter Czerny. 2021. "Characterization of an In Vivo Neutralizing Anti-Vaccinia Virus D8 Single-Chain Fragment Variable (scFv) from a Human Anti-Vaccinia Virus-Specific Recombinant Library" Vaccines 9, no. 11: 1308. https://doi.org/10.3390/vaccines9111308